近期,bat365在线唯一官网登录施建宇教授课题组在药学领域顶级综述类期刊Drug Discovery Today(中科院一区,IF=7.851,年发文量227篇)上发表了题为“Compound–protein interaction prediction by deep learning: Databases, descriptors and models”的综述性论文。bat365在线唯一官网登录博士生杜冰雪为该论文的第一作者,施建宇教授和计算机学院于会副教授为该论文的共同通讯作者。该研究成果得到了国家自然科学基金和陕西省重点研发计划等科研项目的资助。
下文简要介绍了本文的主要内容。
1.研究背景
一种新型小分子药物的开发通常从生物学家确定疾病靶标开始。然后,通过筛选技术在数以万计甚至数以百万计的小分子中挖掘出一套能够抑制或激活该靶点的活性分子。然而,由于大量的高成本试验与高失败率,一个新药的完整开发过程一般要花费10-20年及5亿美元到26亿美元不等的投资。筛选“化合物-蛋白质”相互作用(CPI)是寻找先导化合物的最关键步骤之一(图1)。高通量筛选(HTS)和虚拟筛选(VS)是获得苗头化合物的两种传统方法。然而,传统方法存在固有的局限性,例如小尺寸的化学筛选库,难以获得高质量的三维结构等。基于深度学习的CPI预测方法不仅能够克服传统方法的局限性,而且能够大幅度减少筛选成本。本论文对基于深度学习的CPI预测方法进行了全面调查。
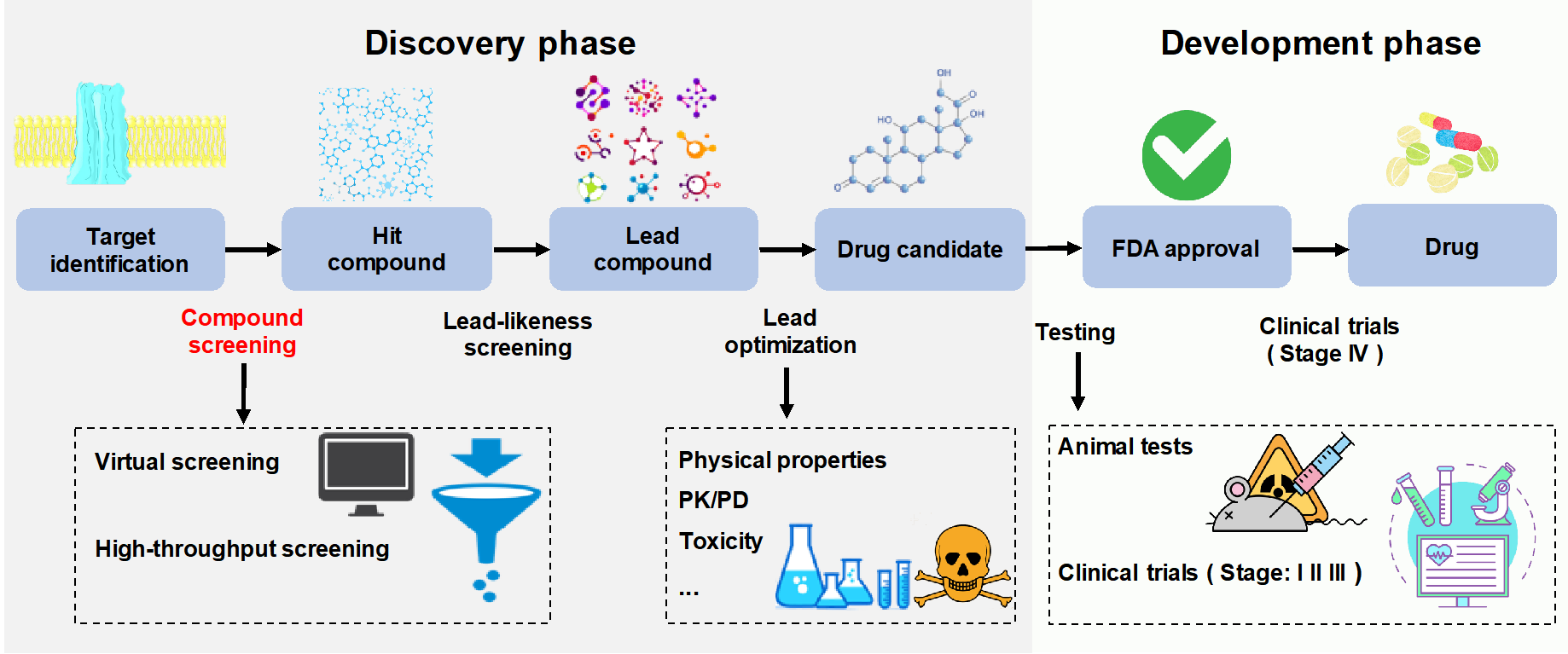
图1 药物设计与开发总体过程
2.数据库及特征表示
其次,本文总结了化合物、蛋白质和绑定复合物的流行数据库:STITCH、BindingDB、PDBbind、BindMoad、KIBA、Davis、DUD-E、KEGG及DrugBank等。
对于机器学习而言,首要步骤始终是对象表示。对于CPI预测来说,对象表示是指将化合物和蛋白质编码为高维特征向量,其中每个维度都反映了化合物和蛋白质的特定属性。化合物描述符包括基于1维,2维及3维的结构表示。常见的表示是基于分子指纹的定性描述符,主要包括:基于子结构、基于路径、圆形、基于药效团以及复合分子指纹(图2)。常见的蛋白质描述符主要包括基于序列和基于结构的描述符。
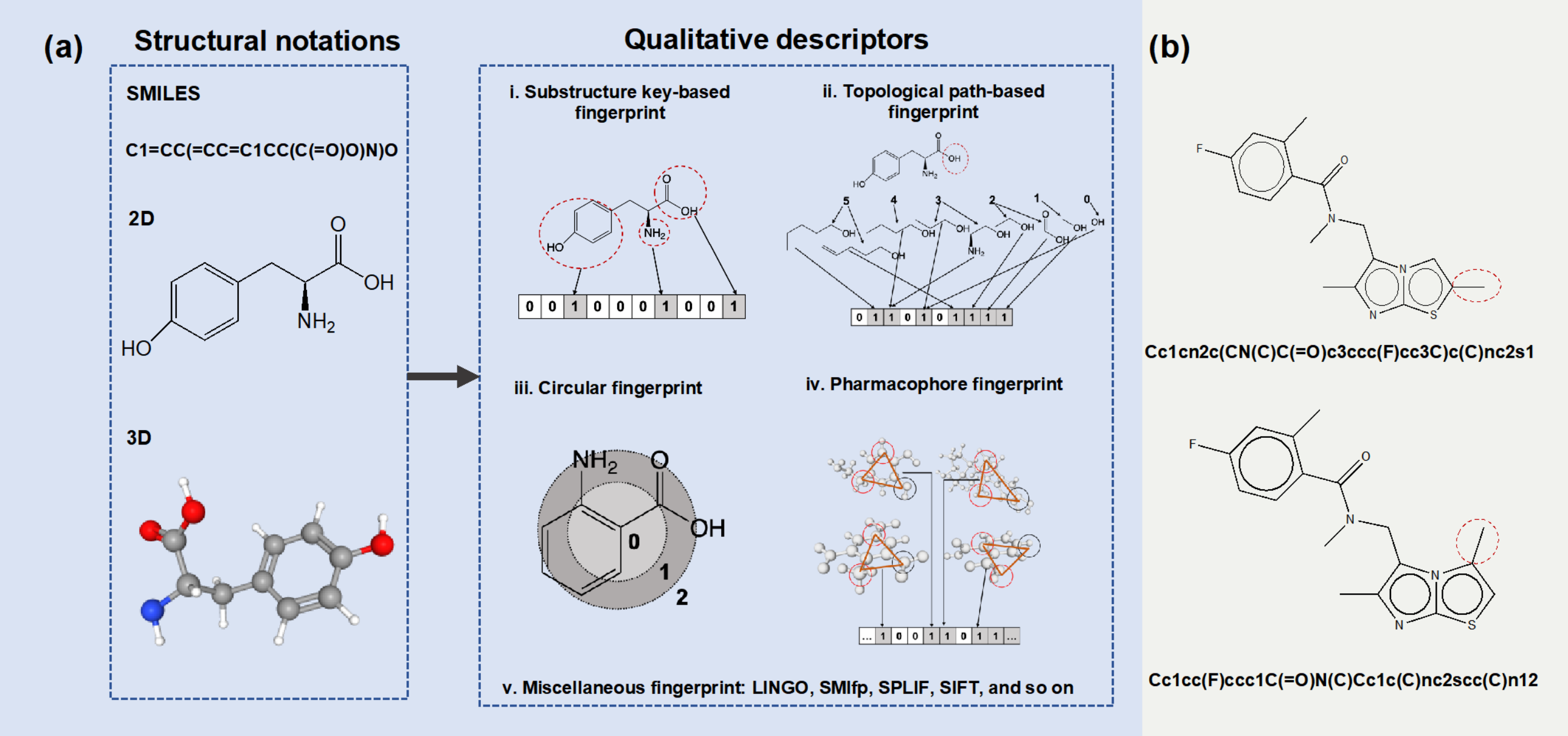
图2 小分子结构表示
3.基于深度学习的CPI预测模型
进而,本文从设计范式的角度简要介绍了最先进的29种基于深度学习的预测模型,包括11种经典模型、9种基于注意力机制的模型以及9种基于复合物的模型。
(1)经典模型是一个Y型框架,其中一个分支实现化合物编码,另一个实现蛋白质编码,化合物和蛋白质的嵌入表示在拼接之后被进一步输入到一个多层深度神经网络,以输出两者的相互作用的置信度或绑定强度。尽管经典模型实现了较好的CPI预测性能,但它们无法明确指出哪些因素对形成相互作用的贡献。由于注意力机制能够在一定程度上揭开深度学习“黑箱”,将注意力层整合到经典“Y”型框架模型中有利于解释化合物与蛋白质形成相互作用的原因。
(2)基于注意力机制的模型通过双向注意力模块或联合注意力模块揭示了对结合起关键作用的蛋白质残基和化合物原子或绑定位点。但是,当前基于注意力机制的可解释性仅通过一个或几个示例进行评估,并且在大规模数据集中是不一致的。此外,缺乏统一的标准来系统地评估各种基于注意力的模型的可解释性。因此,应该设计更合理的注意力层并形成统一的评估指标,以便挖掘化合物和蛋白质的结合规则。
(3)通常大量的蛋白质的结构是很难获取的,但当有蛋白结构或化合物-蛋白质复合物时,设计基于复合物的模型有助于CPI预测的问题。但是基于复合物的模型需要大量计算成本,且与经典模型或基于注意力机制的模型相比并没有显著改进。一种可能的解决方案是获得更多的数据,而另一种是开发基于迁移学习的模型,以将复合物表示从序列模型迁移到3维结构型。
在总结了这些模型的实际理念和优缺点之后,本论文分别在DUD-E和DAVIS两个标准数据集上就“化合物和蛋白质是否绑定”和 “化合物和蛋白质的绑定亲和力大小”两个问题对比分析它们的预测性能。
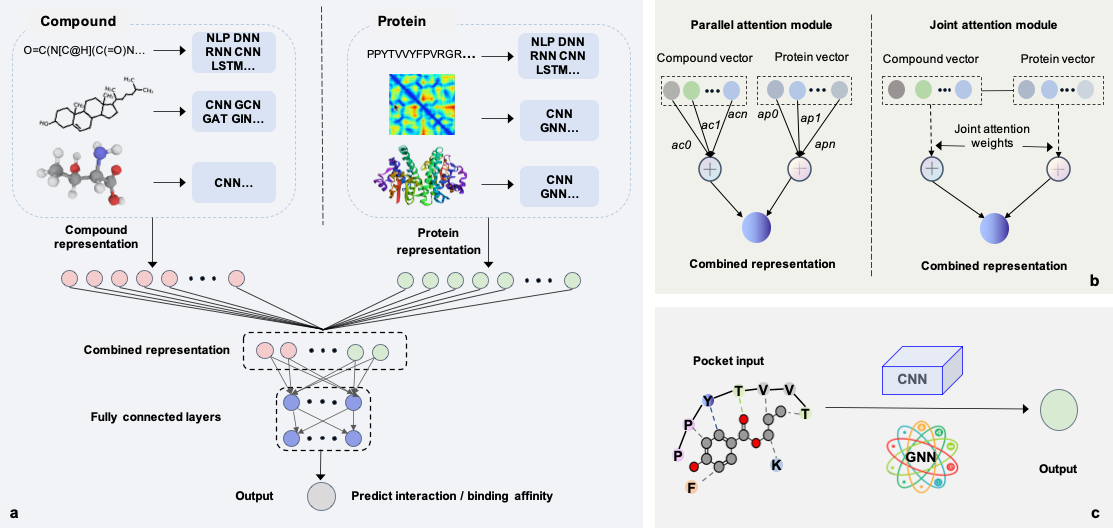
图3 基于深度学习的CPI预测模型构架
4.实际应用
随着人工智能的快速发展,基于深度学习的小分子药物研发也取得重大突破且部分候选药物已进入临床试验。本文列出了近年来的一个典型案例。Insilico Medicine和药明康德在2019年通过深度学习模型,仅在23天就产生了6个先导化合物,并在46天内从中筛选出1个具备良好药代动力学行为的候选药物,最后通过实验验证了其对于效DDR1激酶的高效抑制作用。
由于深度学习探索广阔化学空间的能力,基于深度学习的模型、工具和方法能够在开放和大数据时代加速药物设计。快速发现新的活性化合物并产生新的苗头和先导化合物的成功将推动药物发现的发展。这篇综述不仅有助于启发学术界新模型的设计,还有助于先导化合物的筛选。
文章链接:https://doi.org/10.1016/j.drudis.2022.02.023。(文/图:杜冰雪,审核:杨慧)